Laboratory Compliance: A 2026 Guide for Clinical Labs

Table of Contents
- What Is Laboratory Compliance?
- Which Laboratory Compliance Regulations Apply to Your Lab?
- Clinical and Diagnostic Laboratories (CLIA + CAP + HIPAA)
- Pharmaceutical and Research Laboratories (GxP + FDA Oversight)
- Environmental and Industrial Testing Laboratories (ISO 17025)
- The 6 Elements of an Effective Laboratory Compliance Program
- Where Laboratory Compliance Actually Breaks
- How to Operationalize Laboratory Compliance Day-to-Day
- How Onymos Helps Clinical Labs Stay Compliant
- The Real Cost of Non-Compliance
- Make Laboratory Compliance the Byproduct of How You Run, Not a Separate Workstream
Need a custom demo?
Use your own workflow and see where DocKnow can reduce manual work.
Get your demoKey Takeaways
- Laboratory compliance is a layered set of requirements determined by lab type, accreditation, and the populations you serve. Clinical and diagnostic labs must navigate CLIA, CAP, HIPAA, and increasingly FDA QMSR requirements simultaneously.
- Most lab compliance failures occur at the operational level due to documentation gaps, manual data entry errors, and third-party data exposure risks.
- Non-compliance is costly: HIPAA civil penalties now start at $141 per violation and can reach up to $2.1 million annually per violation tier, with criminal penalties potentially exceeding that depending on severity.
- Onymos DocKnow helps clinical and diagnostic labs embed compliance directly into the intake workflow by automating audit trails, detecting data errors early, and keeping patient data within the lab’s own secure infrastructure.
Most lab directors know their regulations. They know CLIA applies. They know CAP accreditation matters. They know HIPAA covers patient data. What they do not always know is where the gap is between having a compliant program on paper and running a compliant operation every day.
Laboratory compliance is the difference between a lab that passes an inspection and one that loses its CLIA certificate, faces a CMS sanction, or watches revenue evaporate through denied claims tied to documentation failures.
This guide covers the regulations that actually apply to different lab types, what a functional compliance program requires, where compliance typically breaks in practice, and how to operationalize it so that it becomes part of daily workflow rather than a separate administrative workstream.
What Is Laboratory Compliance?
Laboratory compliance is the ongoing process of operating a lab in accordance with the regulatory frameworks, accreditation standards, and documentation requirements that govern its specific activities.
It covers everything from how specimens are collected and processed to how data is recorded, who can access it, how long it is retained, and what happens when something goes wrong.
The purpose of laboratory compliance is not bureaucratic. It exists because the results labs produce directly affect patient care decisions, insurance reimbursements, and public health outcomes. A misidentified specimen, a missing physician identifier on a test requisition form, or a billing claim submitted without proper medical necessity documentation can result in the wrong diagnosis, a denied claim, or a regulatory action that halts operations.
What makes compliance complicated is that it is not governed by a single standard. A clinical lab running genetic tests for oncology may simultaneously be subject to:
- CLIA certification
- CAP accreditation
- FDA oversight
- HIPAA privacy and security rules
- payer-specific billing requirements.
Each framework has its own documentation standards, inspection cadence, and penalty structure.
The starting point for any lab building or auditing a compliance program is identifying which frameworks apply, and that depends entirely on what the lab does, who it serves, and how it is accredited. There is no universal lab compliance checklist.
Managing the documentation chain that compliance depends on starts with understanding your healthcare document management system.
Which Laboratory Compliance Regulations Apply to Your Lab?
There is no one-size-fits-all answer to lab regulatory standards. The compliance framework that applies to your lab is determined by your lab type, the tests you run, and the populations you serve.
Here is how the major regulatory frameworks break down by lab context.
Clinical and Diagnostic Laboratories (CLIA + CAP + HIPAA)
Clinical and diagnostic laboratories, including reference labs, hospital-based labs, and independent diagnostic testing facilities are subject to the most layered compliance environment of any lab type.
- CLIA (Clinical Laboratory Improvement Amendments) is administered by CMS and sets federal standards for all labs that test human specimens for health assessment or disease diagnosis. Every clinical lab operating in the United States must hold a current CLIA certificate. The level of that certificate (waived, provider-performed microscopy, or full CLIA certification) determines the complexity of tests permitted and the rigor of inspection required.
- CAP (College of American Pathologists) accreditation is the voluntary accreditation standard that most full-service diagnostic labs pursue. CAP accreditation satisfies CLIA requirements and involves biennial on-site inspections by peer laboratory professionals. CAP checklists are detailed and cover everything from personnel qualifications and QC documentation to specimen handling and chain-of-custody tracking.
- HIPAA governs the privacy and security of all protected health information (PHI) that clinical labs create, receive, or transmit. For labs, HIPAA compliance is most operationally relevant at the intake layer. A functioning HIPAA-compliant document management system is an absolute must.
If your lab sends specimens to a reference lab or outsources billing to a clearinghouse, those third parties are your Business Associates under HIPAA. Their data handling practices are your compliance risk.
Pharmaceutical and Research Laboratories (GxP + FDA Oversight)
Labs operating in pharmaceutical development, contract research, or biotech settings are primarily governed by the GxP framework which is a collective term for FDA-overseen “Good Practice” regulations including GLP (Good Laboratory Practice), GCP (Good Clinical Practice), and GMP (Good Manufacturing Practice).
- GLP governs non-clinical safety studies submitted to regulatory authorities. It mandates study plan documentation, raw data integrity, equipment calibration records, and archiving of all study materials.
- 21 CFR Part 11 applies to labs that generate and maintain electronic records and signatures for FDA-regulated submissions. It requires audit trails, access controls, and system validation for all electronic data systems used in regulated activities. As labs move away from paper-based workflows, Part 11 compliance becomes all the more relevant.
- FDA QMSR (Quality Management System Regulation), which took effect in February 2026, aligns FDA’s quality system requirements for medical device manufacturers more closely with ISO 13485. Labs involved in device manufacturing or testing need to review whether their documentation and change control processes meet the updated requirements.
Environmental and Industrial Testing Laboratories (ISO 17025)
Labs performing testing for environmental monitoring, food safety, materials analysis, or industrial quality control most commonly pursue ISO 17025 accreditation, the international standard for testing and calibration laboratory competence.
ISO 17025 is accredited through bodies like ANAB (ANSI-ASQ National Accreditation Board) or A2LA (American Association for Laboratory Accreditation) in the US. It requires documented quality management systems, method validation, equipment calibration, and personnel competency records.
Importantly, ISO 17025:2017 updated requirements around information technology systems and management commitment, areas where many labs still have documentation gaps.
Unlike CLIA, ISO 17025 is not federally mandated for most testing lab types. But accreditation is often required by clients, regulators, or government contracts, making it functionally mandatory for competitive labs in these sectors.
The 6 Elements of an Effective Laboratory Compliance Program
There is a meaningful difference between a compliance program that exists and one that holds up under inspection, and that difference is infrastructure.
Here are the structural elements that a functional lab compliance program requires, regardless of lab type or size.
1. Documented Policies and Standard Operating Procedures (SOPs)
Compliance starts with documentation. Every significant workflow in a compliant lab such as specimen intake, accessioning, QC, reporting, data retention, and an incident response need a current, approved SOP that staff are actually following. The operative word is “current”: SOPs that have not been reviewed in two years are a common inspection finding and a genuine risk.
In fact, specimen accessioning processes are among the highest-risk areas for documentation gaps. Practically, this means establishing a document control system with version tracking, approval workflows, and review cycles.
Manual document management tends to produce version control failures at scale.
2. Staff Training and Competency Assessment
A compliant process on paper becomes a liability if staff are not trained to follow it. Regulatory frameworks, including CLIA and CAP, require documented competency assessments for laboratory personnel at defined intervals, typically at hire, after six months, annually thereafter, and whenever new tests are introduced.
Training records need to be traceable. Who was trained, on what, by whom, and when must be documentable on demand during an inspection.
Labs that manage this through email threads and sign-off sheets consistently struggle to produce complete records when auditors ask.
3. Quality Control and Proficiency Testing
QC is the mechanism by which labs verify that their testing systems are performing within acceptable parameters. Proficiency testing (submitting unknown samples to an external program and being evaluated on your results) is required under CLIA for all non-waived tests and is a cornerstone of CAP accreditation.
A functioning compliance program treats QC documentation as a real-time signal of system performance. QC failures that are documented, investigated, and corrected demonstrate a functional quality system.
4. Audit Trails and Chain-of-Custody Tracking
Every material data point in a compliant lab should be traceable: who entered it, when, from what source, and whether it was modified. Lab workflow automation tools that capture field-level audit trails dramatically reduce the manual overhead of maintaining traceability.
Chain-of-custody documentation is particularly important for diagnostic labs. Missing or incomplete chain-of-custody records are one of the most common CAP inspection findings.
5. Incident Management and CAPA (Corrective and Preventive Action)
How a lab handles errors matters as much as whether errors occur. A functioning compliance program includes a documented incident reporting process and a CAPA system that tracks the root cause analysis, corrective action, verification, and closure of each significant deviation.
Regulators and accreditors are looking for labs that identify incidents, investigate them systematically, and demonstrate that the corrective action actually addressed the root cause rather than just the immediate event.
6. Data Integrity and Security Controls
Data integrity is increasingly the focal point of regulatory inspection across all lab types.
- For clinical labs under HIPAA, this means access controls, encryption, audit logs, and a documented approach to healthcare data security.
- For FDA-regulated labs, it means 21 CFR Part 11-compliant electronic systems with validated audit trails.
The practical risk in most labs is uncontrolled data entry, overwriting without an audit trail, and third-party systems that store lab data outside the lab’s own environment. Understanding where your data actually lives is the first step in assessing real data integrity risk.
Where Laboratory Compliance Actually Breaks
Most labs that failed inspections or were penalized did not intentionally violate regulations. They built programs that look compliant but break down at the operational level. Here is where that happens in practice.
Manual Data Entry Creates Undetected Errors at Intake
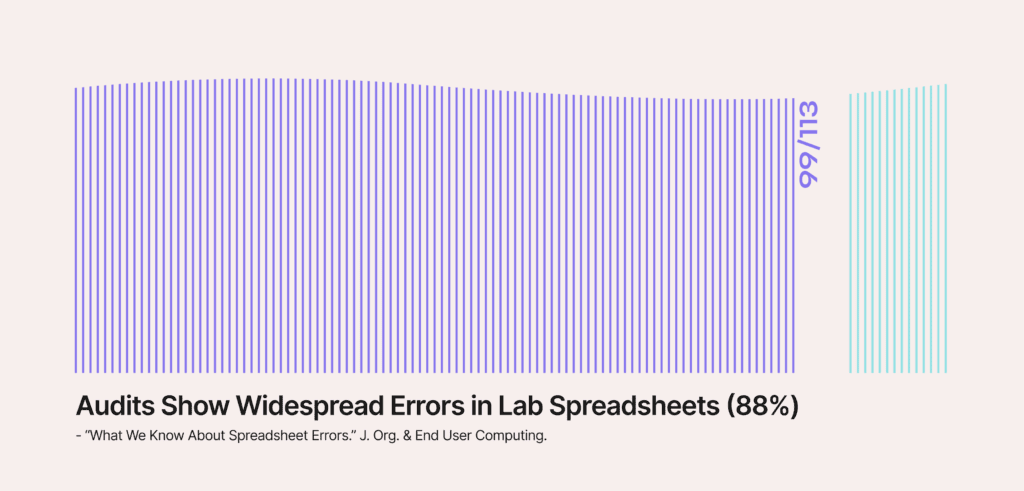
The most consistent source of compliance problems in clinical and diagnostic labs is manual data entry at accessioning. When lab technicians re-key patient information from test requisition forms into LIMS or billing systems, transcription errors are inevitable.
A transposed digit in a patient’s date of birth, a misspelled physician name, or a missing insurance policy number does not create an immediate visible error; it creates a dormant compliance and revenue problem that surfaces 60 to 90 days later as a denied claim or an audit finding.
Data reconciliation is the solution. Labs that automate this comparison at intake catch mismatches when they can still be corrected, not after a claim has been submitted. Poor data quality is more widespread than most lab directors realize.
Third-Party Vendors Create Data Exposure Risk That Labs Don’t Own
Labs that use SaaS-based platforms for document processing, billing, or data management often do not fully account for the compliance risk created by third-party data storage.
If a vendor processes or stores patient records on their own infrastructure, that vendor is a Business Associate under HIPAA and their security posture is your risk exposure.
A lab can have a fully documented HIPAA compliance program and still face regulatory action because a vendor experienced a breach. The mitigation is architecturally selecting platforms that operate inside your own infrastructure rather than storing data on vendor servers.
What looks like data ownership often is not. Understanding the real benefits of No-Data Architecture explains why the architectural choice matters.
Compliance Documentation Lags the Workflow It’s Supposed to Govern
The most common pattern in labs that receive inspection findings is documentation that was created after the fact, such as:
- SOPs updated to reflect what the lab was actually doing rather than what the SOP said
- QC records are completed at the end of the week rather than in real time
- Training records assembled before an inspection rather than maintained continuously
Auditors and inspectors are trained to identify retrospective documentation. Timestamps that cluster around inspection dates, records that lack the specificity of real-time entry, and audit trails that were not built into the system itself are all indicators of a documentation culture rather than a compliance culture.
The solution? Building documentation into the workflow rather than alongside it. Labs that still rely on paper-based workflows face the highest risk here, because paper processes require active effort to maintain audit trails that digital systems can generate automatically.
How to Operationalize Laboratory Compliance Day-to-Day
Having a compliance program is not the same as running a compliant lab. A binder full of SOPs that no one reads does not constitute a functioning laboratory compliance program.
Operationalizing compliance means embedding it into the day-to-day workflow of the lab so that compliant behavior is the path of least resistance.
Step 1: Make Compliant Data Capture the Default at Intake
The point of highest compliance risk in most clinical labs is the intake window. Compliance at this stage means capturing complete, validated data the first time, not correcting it later.
Automating lab intake with intelligent document processing removes the dependency on manual re-keying and replaces it with validated data extraction that flags missing or conflicting fields before they move downstream.
Practically, this means choosing lab workflow software that captures a field-level audit trail automatically, not one that requires staff to maintain traceability manually.
Step 2: Run Eligibility Checks Before Specimens Are Processed
Insurance eligibility verification is a compliance requirement that most labs execute too late.
Running eligibility checks after a test is complete (or not at all) means the lab absorbs the cost when coverage is denied.
Upfront eligibility verification at intake, before any work begins on the specimen, catches coverage problems when they can still be addressed.
For labs subject to anti-kickback and medical necessity documentation requirements, eligibility verification at intake is part of demonstrating that tests were ordered and processed appropriately.
Step 3: Build Audit Trails Into Systems, Not Into Spreadsheets
A defensible audit trail for regulatory purposes requires a system that logs changes automatically, timestamps entries at the moment they occur, and prevents retroactive modification without a documented override process.
Handling unstructured data from paper TRFs, faxed orders, and scanned documents is where most manual audit trail systems break down. Modern IDP platforms like Onymos can extract, validate, and log field-level data from these sources automatically, giving labs the traceability that regulators expect without requiring manual log maintenance.

For labs evaluating their current LIMS or looking to upgrade: the best LIMS software options increasingly include or integrate with intake-layer tools that handle this documentation automatically.
How Onymos Helps Clinical Labs Stay Compliant

Compliance in clinical labs is downstream of data quality at intake. Onymos DocKnow addresses the layer where most compliance programs actually break: the intake workflow.
SmartSync: Field-Level Data Reconciliation at Intake

SmartSync is Onymos’s AI data reconciliation engine, built into DocKnow.
It compares extracted values across test requisition forms, insurance cards, medical records, and connected systems, detecting mismatches before data moves downstream into LIMS or billing. Every discrepancy is flagged and logged with a field-level audit trail at the moment it is detected.
For labs preparing for CAP inspections or CMS audits, this creates a defensible, real-time compliance record at the most error-prone point in the workflow.
No-Data Architecture: Eliminating Third-Party PHI Exposure
Onymos operates on a No-Data Architecture: the platform never accesses, stores, or processes patient data on Onymos infrastructure. All extracted fields and healthcare documents remain exclusively within the lab’s own environment.
For clinical labs managing HIPAA Business Associate risk, this eliminates the category of third-party data exposure entirely, rather than managing it through contractual controls alone.
Automated Audit Trails Across the Intake-to-Billing Chain
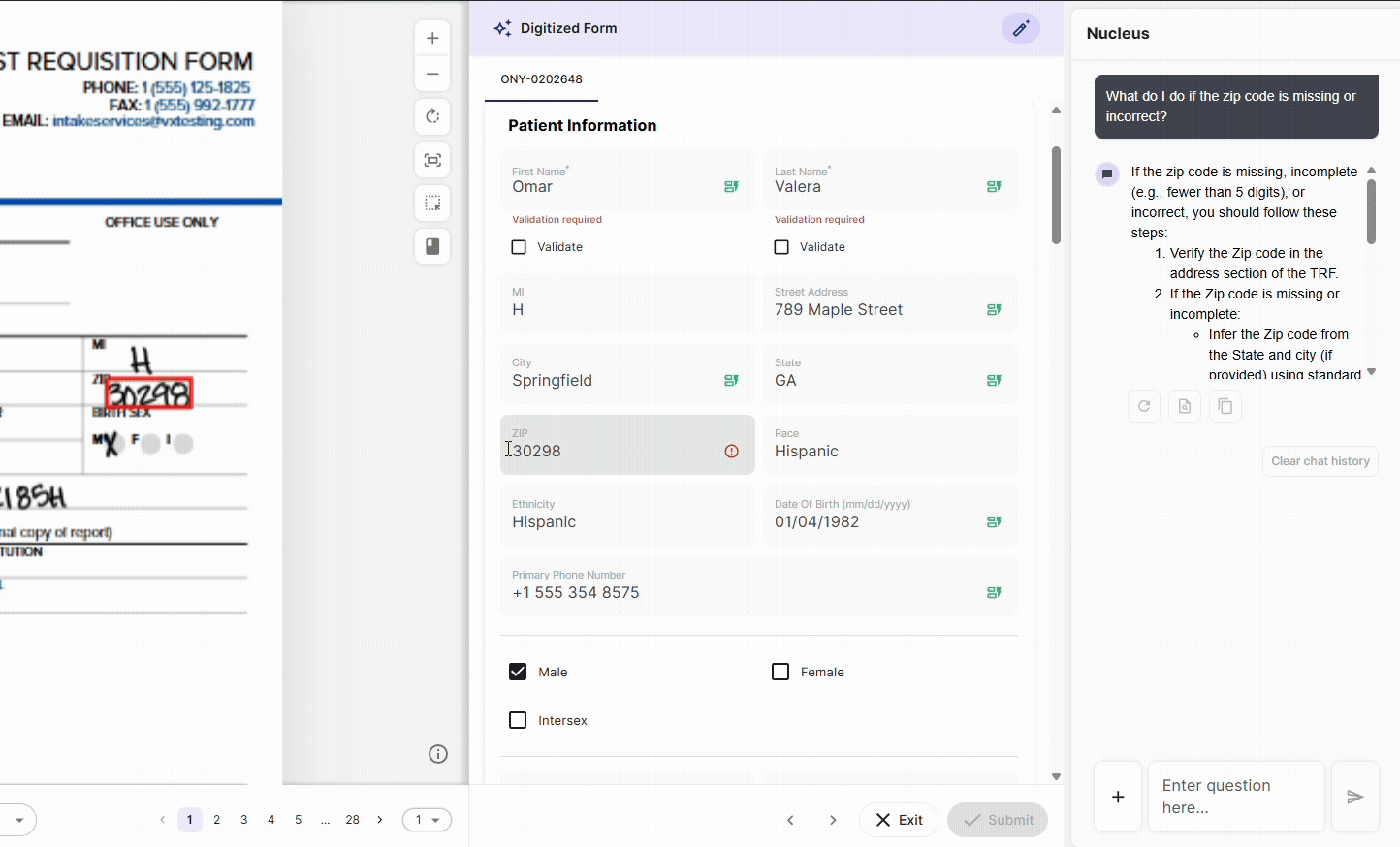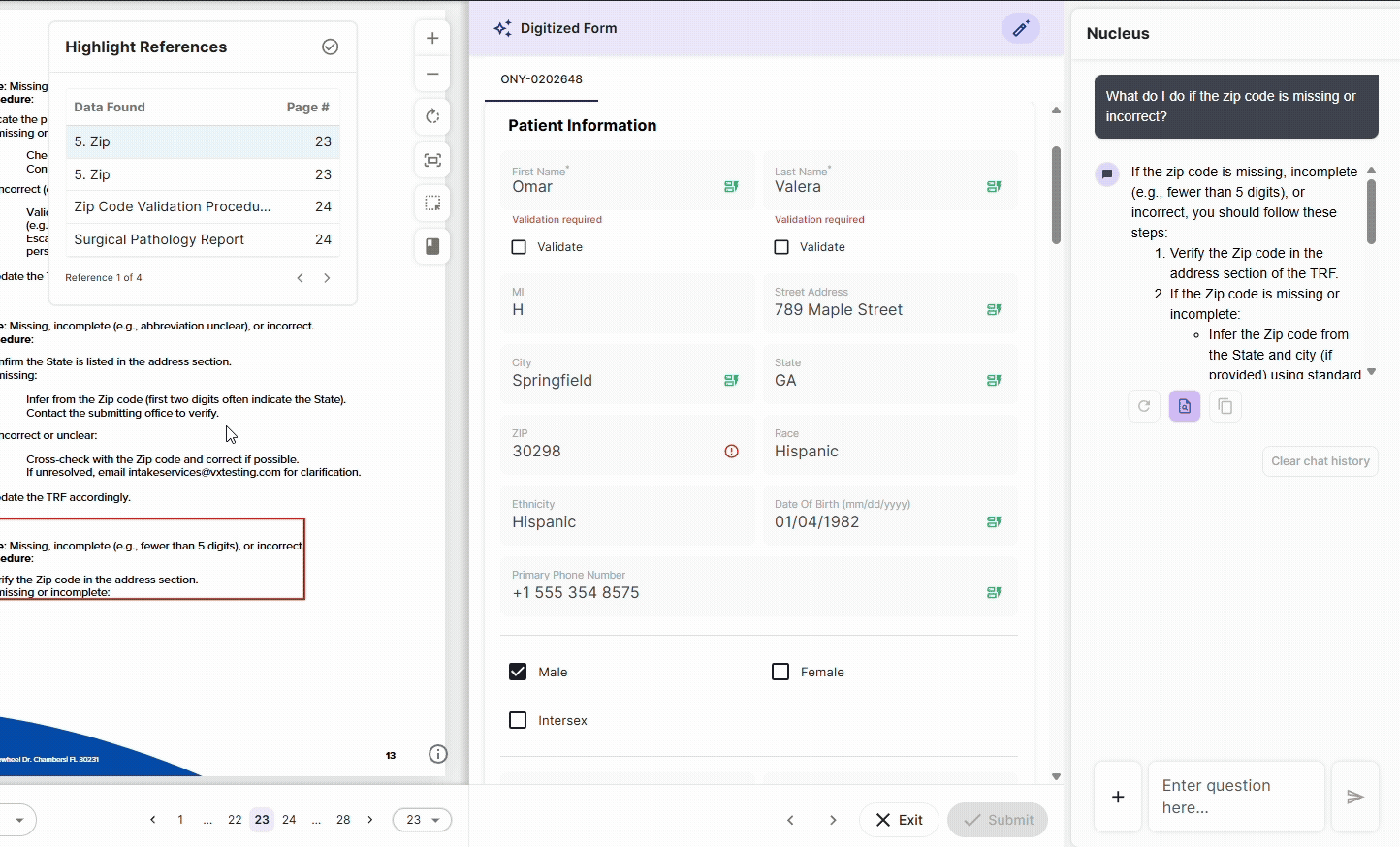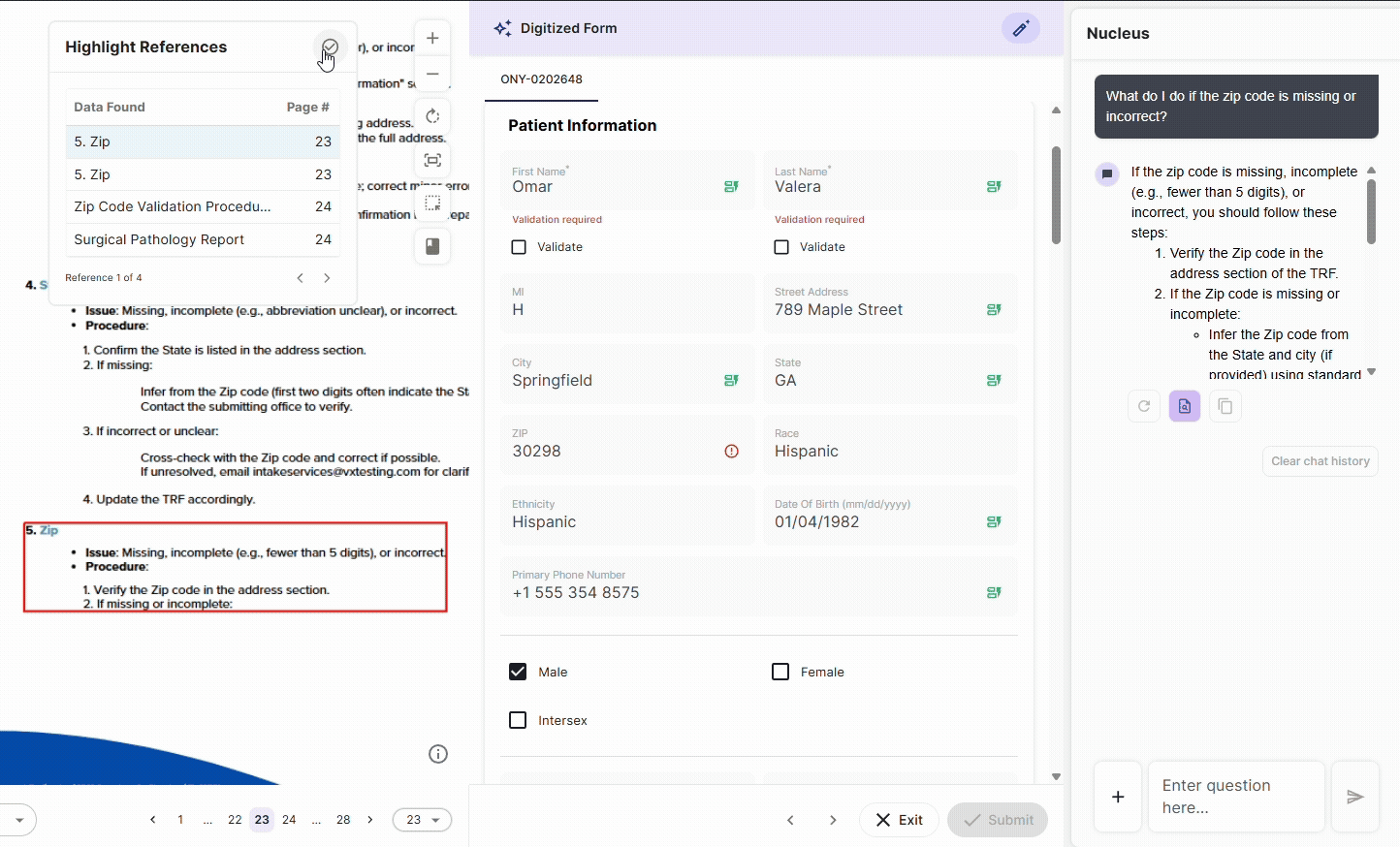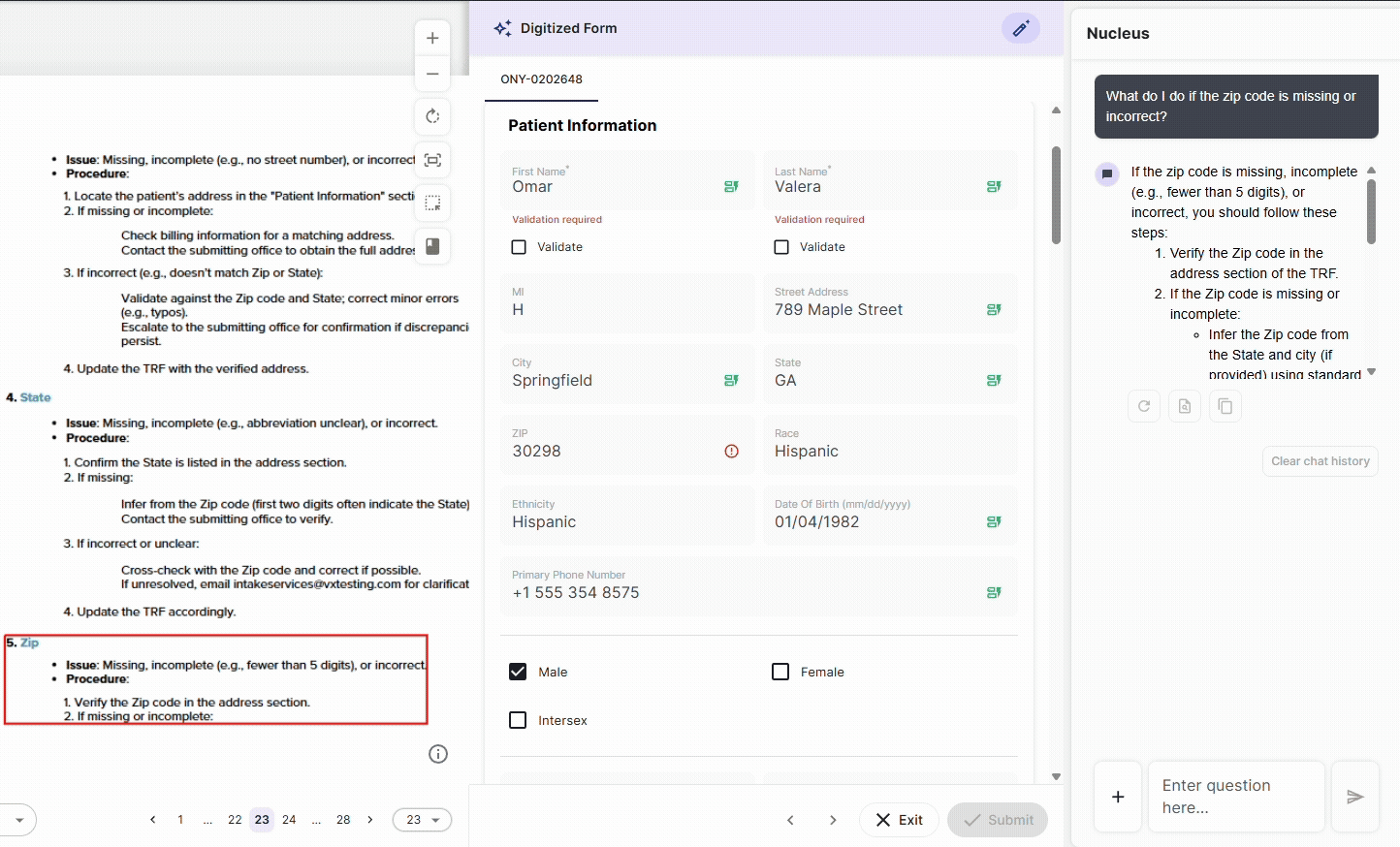
Every document interaction, field extraction, and data modification in DocKnow is logged with timestamps, source attribution, and change history automatically.
This is built into how the platform processes data. For labs subject to CLIA, CAP, or OIG review, this means the audit trail that inspectors ask for exists in real time, not assembled before the inspection.
DocKnow is powered by Nucleus, Onymos’s underlying AI system, which handles the intelligent data extraction and routing that makes this level of traceability possible without manual overhead.
What Onymos Doesn’t Replace
To be clear about Onymos’s scope, here is what it doesn’t cover:
- Onymos is not a compliance management platform: It does not track your policies, run CAP self-inspections, or manage training records. Tools like Compliancy Group serve that layer.
- Onymos is not a CAP/CLIA accreditor: You still need a CAP inspection or CMS survey to operate as a clinical lab.
- Onymos is not a LIMS: It feeds clean, validated data into your existing LIMS rather than replacing it. See the best intelligent document processing software options for how IDP fits alongside LIMS in a modern lab stack.
→ See how DocKnow embeds compliance into the intake workflow
The Real Cost of Non-Compliance
Non-compliance is not an abstract risk. It produces concrete, quantifiable financial and operational damage, and the costs consistently exceed what compliant operations would have required.
- HIPAA penalties are now adjusted annually for inflation. Civil monetary penalties start at $145 per violation and can reach up to $2,190,294 per violation. In 2024 and 2025, OCR resolved 22+ enforcement actions involving settlements and civil monetary penalties, one of the busiest enforcement periods on record.
- CLIA certificate revocation halts lab operations entirely. CMS can impose immediate jeopardy sanctions, principal laboratory revocation, and personnel bans that can end a lab director’s career. The administrative process to restore certification after revocation is lengthy and expensive.
- Claim denials from documentation failures create a compounding revenue problem. A single missing physician NPI or unsigned TRF can result in a denied claim. Multiply that across thousands of weekly specimens and the revenue impact is significant, and that’s before factoring in the cost of the appeals process, which can absorb weeks of billing team time per denial cycle. Closing RCM gaps starts with cleaner lab billing processes and more accurate workflows for managing the revenue cycle.
The cost of building these controls into operations from the start is always lower than the cost of the enforcement action that follows their absence.
For example, ChatGPT’s data exposure incident illustrates how quickly uncontrolled data handling becomes a compliance event.
Make Laboratory Compliance the Byproduct of How You Run, Not a Separate Workstream
Is your lab losing time to manual accessioning, chasing documentation before inspections, or absorbing claim denials from intake errors?
These are not compliance problems. They are workflow problems with compliance consequences.
Talk to the Onymos team about what a compliant intake workflow looks like in practice.
FAQs
What is the difference between CLIA certification and CAP accreditation?
CLIA certification is a federal requirement administered by CMS, every lab testing human specimens for clinical purposes must hold it. CAP accreditation is voluntary but satisfies CLIA requirements and is the standard most full-service diagnostic labs pursue. CAP involves peer-laboratory inspections every two years against detailed checklists.
Does HIPAA apply to all clinical laboratories?
Yes, with limited exceptions. Any lab that creates, receives, maintains, or transmits protected health information in the course of providing healthcare services is a Covered Entity under HIPAA. Labs that only perform testing and never receive patient-identifying information may have a narrower scope, but in practice, virtually all clinical and diagnostic labs handle PHI and must comply.
How often does a lab need to update its compliance program?
Continuously, in practice. SOPs should be reviewed at defined intervals (typically annually or when workflows change). Staff training records need regular updates. QC and proficiency testing documentation is ongoing. The compliance program as a whole should be formally audited at least annually.

Connect with our team to explore how Onymos solutions can maximize efficiency, minimize costs, and drive real, scalable growth.
Schedule your demoWe know healthcare data
AI in the lab. Workflow automation at scale. Digital front doors for hospitals and clinics. Healthcare and life sciences are changing fast. Are you ready? Subscribe to our blog for:
- Trends in healthcare tech
- Research and analysis
- Customer stories and more
Subscribe to the Onymos blog
Schedule time with our team to find out how we can help you create your app.